
 |
 |
 |
 |
 |
Artigo
| Stereoselective oxidative coupling of methyl trans-ferulate to (±)-trans-dehydrodiferulate dimethyl ester by Trametes versicolor FC: an enantioselective green approach |
|
Eliane O. SilvaI,* I. Departamento de Química Orgânica, Instituto de Química, Universidade Federal da Bahia, 40170-115 Salvador - BA, Brasil Received: 01/30/2025 *e-mail: elianeos@ufba.br Chemoenzymatic strategies to synthesize complex molecules have been developed by combining biocatalytic and synthetic methods in a multistep approach toward a target molecule. Dihydrobenzofuran neolignans (DBNs), a class of plant-derived natural compounds, display several biological activities. Oxidative coupling of phenylpropanoids is commonly used to obtain DBNs, but yields are low, and stereochemical control is poor. This study reports the novel eco-friendly and stereoselective conversion of methyl trans-ferulate (1) into DBN 2 ((±)-trans-dehydrodiferulate dimethyl ester) promoted by Trametes versicolor FC in 47% yield. Compared to Ag2O-promoted oxidative coupling, this biotransformation approach is enantioselective (66.7% ee of the main enantiomer) and affords DBN 2, as confirmed by chiral-HPLC (high-performance liquid chromatography) and NMR (nuclear magnetic resonance) data analyses. Our preliminary results highlighted the combination of biotransformation and traditional chemical processes as a promising starting point for obtaining DBNs from the corresponding phenylpropanoids enantioselectively. INTRODUCTION Dihydrobenzofuran neolignans (DBNs) are phenolic compounds bearing two phenylpropanoid (C6C3) units joined through C8–C5' and C7–O4' bonds.1 These compounds display several biological and pharmacological activities, such as trypanosomal,2 insecticidal,3 schistosomicidal,4 antileishmanial,5 antitumor,6,7 and anti-inflammatory action.8 Although DBNs are structurally diverse, their biological evaluations have been limited due to low yields obtained from traditional phytochemical isolation methods.9 Many strategies have been proposed to synthesize DBNs. Most of these strategies are based on the oxidative coupling of phenylpropanoid units, which mimics their biosynthesis in vascular plants.10,11 The most common protocol starts with silver(I) oxide (Ag2O) to promote oxidative coupling of phenylpropanoids.12 The reaction is diastereoselective and gives DBNs as racemic mixtures of trans-enantiomers in 20-40% yield.13 Many efforts have been dedicated to replace Ag2O in achieving DBNs with higher stereoselectivity and yields. For instance, iron salts,14 Ru and Rh complexes15 have been employed, but the stereoselectivities of the coupling reactions that conduct to DBNs have not been clearly described. Methyl trans-ferulate (1) may be synthetically converted into the DBN 2 (Scheme 1), which is the key intermediate for the synthesis of 3',4-di-O-methylcedrusin,16 one of the compounds underlying the healing properties of "dragon's blood", a latex produced by some Croton sp. (Euphorbiacae) from South America.17 However, the standard protocol (Scheme 1a) that is used to synthesize DBN 2 requires toxic reactants and leads to racemic mixtures of (R,S)- and (S,R)-enantiomers in low yields, along with toxic by-products and other coupled products.12
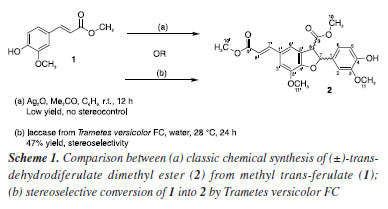
Innovative and eco-friendly strategies to synthesize bioactive natural products have been continuously studied and developed.18,19 In this scenario, chemoenzymatic strategies hold enormous potential for streamlining access to important bioactive molecules. Organic chemists have explored the chemoenzymatic approach by combining biocatalytic and synthetic organic methods to obtain interesting small bioactive molecules.20 Indeed, the broad enzymatic potential of fungi has ensured that they play an essential role in biotechnology studies focused on developing new selective chemical reactions.21 For instance, enzymes such as horseradish peroxidase have been assayed as biocatalysts to produce DBNs scaffold with good enantioselectivity.22 Nevertheless, the use of pure enzymes has been limited by costs. Whole fungal cells are an acceptable source of enzymes, and they can convert various substrates through specific chemical reactions while efficiently regenerating cofactors.23 Despite some limitations, mainly the toxicity of some substrates and products to the fungal cell, fungi catalyze numerous chemical reactions that are difficult to carry out by synthetic chemistry processes.24 White-rot fungi, such as the Basidiomycete Trametes versicolor, are a renewable source of enzyme systems composed of laccases and high redox peroxidases, including lignin peroxidase and manganese peroxidase.25,26 Laccases, which belong to the class of multicopper oxidases, have non-specific oxidation capacity, catalyze the oxidation of countless aromatic substrates,27 use air as an oxidant agent, and produce water as the only by-product.28 Among other applications, laccases have been successfully employed to oxidize phenolic compounds, and the resulting radicals undergo oxidative coupling.29 We previously demonstrated through in vitro and in silico evidence the selective capabilities of laccases produced by T. versicolor in selective biotransformation of methyl p-coumarate, methyl ferulate, and methyl caffeate.30 Now, as part of our ongoing interest in obtaining biologically active DBNs,2-4,10,31 this study aims to screen a Trametes versicolor strain isolated from a soil sample for regio- and enantioselective coupling of methyl trans-ferulate (1) to produce trans-dehydrodiferulate dimethyl ester (2) (Scheme 1b). Our results highlight the stereoselectivity of the biotransformation of 1 into DBN 2 by Trametes versicolor FC. Compared to Ag2O-promoted oxidative coupling, our developed biotransformation approach is enantioselective and affords DBN 2 at good yield (47%).
EXPERIMENTAL General procedures and chromatographic analysis The crude biotransformation extract was initially analyzed by thin layer chromatography (TLC, Merck, Germany); ethyl-acetate and hexane (1:1, v/v) were used as mobile phase. Next, high-performance liquid chromatography (HPLC) analyses were carried out on a system (Shimadzu, Kyoto, Japan) equipped with an LC-20AT solvent pump unit, a DGU-20A5 online degassing unit, a CTO-20A column oven kept at 25 °C, an SPD-M20A photodiode array detector operating at 254 nm, and a CBM-20A system controller. Analytical and preparative HPLC analyses of the crude biotransformation extract were carried out on Phenomenex (Torrance, CA) Luna 5µ C18 100 Å 250 × 4.6 mm and 250 × 21.2 mm columns, respectively, using a gradient elution of MeOH:H2O from 65:35 (v/v) to 95:5 (v/v) in 35 min. The biotransformation product was identified on the basis of 1H and 13C nuclear magnetic resonance (NMR) and electrospray ionization mass spectrometry (ESI-MS) data (see Supplementary Material) and by comparison with literature data.32 1H and 13C NMR spectra were recorded on a Bruker Avance DRX400 spectrometer (Karlsruhe, Germany, 400 MHz for 1H and 100 MHz for 13C). Chemical shifts (d) were referenced to the residual deuterated acetone (acetone-d6) peak at dH 2.05 for 1H or 206.3 for 13C. Mass spectrum was recorded on a micrO-TOF-QII mass spectrometer (Bruker Daltonics, Billerica, MA, USA) fitted with an ESI source operating in the negative ion mode and equipped with a hybrid quadrupole time-of-flight (QTOF) mass analyzer. The samples were dissolved in methanol-water (9:1 v/v) and directly infused into the ionization source at a flow rate of 300 μL min–1. Chiral separations were performed on a Chiralpack AD-H chiral column (150 × 4.6 mm ID, 5 µm particle size, Daicel Chiral Technologies, Shanghai, China); hexane/isopropanol 9:1 (v/v) at a flow rate of 1 mL min–1 was employed as mobile phase, and the isocratic elution mode was used. The LC Solution software version 1.25 SP1 (Shimadzu, Kyoto, Japan) helped to control the instrument and to acquire data. To establish the chromatographic analytical method and to compare the selectivity of the synthetic and biotransformation approaches, methyl trans-ferulate (Sigma-Aldrich, St. Louis, MO, USA, 98% purity) was submitted to oxidative coupling with Ag2O (Sigma-Aldrich) as the oxidant agent in a mixture of acetone and benzene (5:8, v/v) placed in a two-neck flask with aluminum foil, on a magnetic stirrer, coupled to a gas tube of N2; the reaction was conducted at room temperature for 20 h, as previously reported.32 The product was purified by column chromatography (silica gel 60, 0.040-0.063 mm), and hexane/ethyl acetate (2:1, v/v) was used as eluent. The synthesis product (43% yield) consisted of a mixture of trans-enantiomers. The stereoselectivity of each reaction was determined by using normalized peak areas without a correction factor. (±)-trans-Dehydrodiferulate dimethyl ester (2) Yellow oil, 80.0 mg, 47 % yield; 1H NMR (400 MHz, acetone-d6, Figure 1S, Supplementary Material) d 3.73 (3H, s, H10), 3.81 (3H, s, H10'), 3.84 (3H, s, H11), 3.92 (3H, s, H11'), 4.47 (1H, d, J3,2 8.1 Hz, H8), 6.04 (1H, d, J2,3 8.1 Hz, H7), 6.44 (1H, d, J9,8 16.1 Hz, H8'), 6.84 (1H, d, J5',6' 8.1 Hz, H5), 6.92 (1H, dd, J6',5' 8.1 Hz, J6',2' 1.7 Hz, H6), 7.10 (1H, d, J2',6' 1.7 Hz, H2), 7.29 (1H, br s, H7'), 7.33 (1H, br s, H6'), 7.63 (1H, d, J8,9 16.1 Hz, H7'); 13C NMR (100 MHz, acetone-d6, Figure 2S, Supplementary Material) d 52.1 (CH3, C10'), 53.5 (CH3, C10), 56.4 (CH3, C11), 56.8 (CH3, C11'), 57.0 (CH, C8), 88.8 (CH, C7), 111.2 (CH, C2), 113.9 (CH, C2'), 116.3 (CH, C5), 116.8 (CH, C8'), 119.5 (CH, C6'), 120.7 (CH, C6), 127.8 (C, C5'), 129.9 (C, C1'), 132.5 (C, C1), 145.9 (CH, C7'), 146.3 (C, C3'), 148.5 (C, C4), 149.1 (C, C3), 151.5 (C, C4'), 168.2 (C, C9'), 172.1 (C=O, C9); HRMS (ESI) m/z, calcd. for C22H21O8 [M - H]–: 413.1242, found: 413.1241 (Figure 4S, Supplementary Material, error 2.4 ppm). Screening for stereoselective oxidative coupling of methyl trans-ferulate (1) by Trametes versicolor FC For the initial biotransformation screening, the fungal strain was cultured in Petri dishes containing potato dextrose agar (PDA, Kasvi) at 28 °C for seven days. The resulting cultures were used in the biotransformation assays. To this end, an initial inoculum of ten 5-mm disks containing mycelia and agar (780 mg) was added to 250-mL Erlenmeyer flasks holding 100 mL of a fermentative medium consisting of 0.20% malt extract (Himedia, Mumbai, India), 0.20% glucose (CAAL, Jaguaribe, SP, Brazil), 0.10% peptone (Himedia, Mumbai, India), 0.10% Tween 80 (Proquimios, Bangu, RJ, Brazil), and 0.0005% CuSO4.5H2O (Synth, Diadema, SP, Brazil). Then, methyl trans-ferulate (1, 10 mg) was dissolved in 500 μL of dimethyl sulfoxide (DMSO; Synth, Diadema, SP, Brazil) and added to each Erlenmeyer flask. All the flasks were incubated at 28 °C and 140 rpm for seven days. Three controls were used: (i) one control consisted of the culture medium, DMSO, and fungus, without substrate; (ii) another control consisted of the culture medium and substrate, without fungus; and (iii) the third control consisted of culture medium only. Controls (i), (ii), and (iii) aimed to demonstrate fungal metabolites (not correlated with the biotransformation), the possible transformation of the substrate catalyzed by the culture medium, and the sterility of the culture medium, respectively. All the experiments were performed in triplicate. Samples were taken daily. Mycelia were separated by filtration, and the fermentation broths were extracted with ethyl acetate (Synth). The solvent was evaporated under reduced pressure to yield crude extracts, which were analyzed by TLC and chiral HPLC. The biotransformation was scaled up by using 17 Erlenmeyer flasks containing 170 mg of 1, incubated under the previously described conditions for one day. The ethyl acetate crude extract (320.0 mg) was obtained and purified by flash chromatography over silica, to yield 80.0 mg of (±)-trans-dehydrodiferulate dimethyl ester (2, 47% yield). Sequences and phylogenetic analysis for fungus identification The fungus was identified by sequencing its ITS (internal transcribed spacer) region. To this end, glass microspheres (diameter of 425-600 μm, Sigma-Aldrich) were used to extract the fungus DNA (deoxyribonucleic acid) through physical lysis of the mycelium.33 The ITS region was amplified by employing ITS1/ITS4 primers. The amplification product was purified (Gel band purification Kit, GE Healthcare), and DNA fragment sequencing was carried out on the ABI 3500 XL Series (Applied Biosystems) system. Genetic distance analyses for dendrogram achievement were performed with the partial sequences of the genes assembled in contigs by the BioEdit 7.2.6 software34 and its comparison with reported sequences at GenBank and CBS. CLUSTALX aligned the sequences.35 The phylogenetic tree was constructed by using the MEGA (Molecular Evolutionary Genetics Analysis) 6.0 software.36 The Neighbor-Joining method37 was employed to establish the evolutionary relationship, and distances were calculated with the Kimura 2-parameter model.38 The statistical support of nodes was estimated by bootstrap analysis with 1000 replications.39 The achieved genomic sequence was deposited at the GenBank database under the accession code PP905504. Laccase activity assay At 28 °C, the fungal strain T. versicolor FC was incubated in the same fermentative medium that was used during the biotransformation assays for 24, 48, 72, or 196 h. Next, fungal cultures were filtered, and the laccase activity was spectrophotometrically determined at 415 nm (Quimis Q780U, São Paulo, Brazil) as described previously;40 2,2-azino-di-[3-ethylbenzo-thiazolin-sulfonate] (ABTS) was employed as substrate. The reaction mixture contained 0.5 mM ABTS, 1.0 mL of 0.1 M sodium acetate buffer (pH 5), and fungus. For the quantification, a calibration curve was carried out with ABTS solutions at a concentration range of 0.2 to 1 mM. Laccase oxidizes the redox indicator ABTS to form a blue-green cation radical. Absorbance was read against a suitable blank, and ABTS oxidation was monitored by determining the increase in A420 (ε 420, 3.6 × 104 M–1 cm–1). One activity unit (U) was defined as the amount of enzyme that oxidized 1 mol of ABTS per min at 25 °C, and the activities were expressed in U L–1. All the assays were carried out in triplicate.
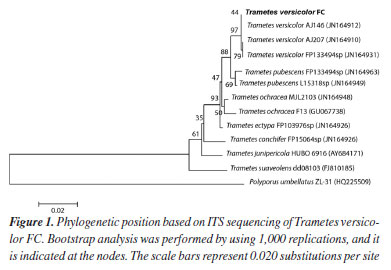
RESULTS AND DISCUSSION Experiments were initiated by developing the culture medium where the fungal strain would have optimal conditions to produce and to secrete laccase, one of the key enzymes for oxidative coupling of methyl trans-ferulate (1). Laccase synthesis and secretion by microorganisms is strictly influenced by nutrient levels, culture conditions, and developmental stage, as well as by the addition of inducers into the culture medium.41 Therefore, biotransformation of 1 by the selected fungus was monitored in a culture medium containing Tween 80 and copper sulfate, which are recognized laccase inducers, for seven days.42 Biotransformation samples were taken daily, and their ethyl acetate crude extracts were achieved. Initially, TLC analysis of the biotransformation crude extract and the synthetic (±)-trans-dehydrodiferulate dimethyl ester (2), used as standard, showed that oxidative coupling of 1 occurred on the first day of incubation. Moreover, TLC analysis showed that the control did not contain DBN 2. Next, the selected fungal strain was identified by sequencing the ITS region, which has been used as the official barcode for the molecular identification of fungi.43 Our soil strain was shown to be closely related and formed a cluster supported by 97% bootstrap value (Figure 1) with Trametes versicolor (100% BLAST (Basic Local Alignment Search Tool) similarity). Therefore, the fungus was identified as Trametes versicolor FC, which belongs to the phylum Basidiomycota. Basidiomycetes are biotechnologically important ligninolytic fungi: they produce laccases44 and have different applications in the biodegradation of phenolic compounds.
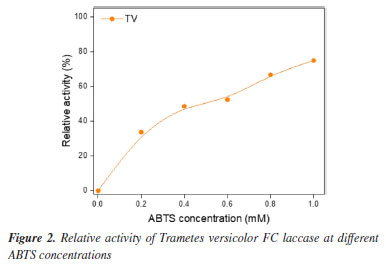
To validate our biotransformation approach as a viable source for in situ laccase production, T. versicolor FC was incubated in the same fermentative medium that was used during biotransformation of 1 for 24, 48, 72, or 196 h. Laccase activity was assayed by using ABTS as substrate (Figure 2). When 1 mM ABTS was reached, 74.9% of relative laccase activity was achieved. The relative activity of laccase increased with increasing ABTS concentration, indicating that higher substrate concentration did not inhibit the reaction. In other words, laccase produced by T. versicolor FC in the culture medium employed in the biotransformation of 1 had efficient potential even at higher ABTS concentrations. Given that Trametes genus is a traditional source of laccase,45 biotransformation of 1 into DBN 2 may have been catalyzed by laccase. The use of inducers of laccases in the culture medium where the biotransformation was carried out, along with the detected laccase activity, reinforced the hypothesis that laccase underlay the biotransformation reaction investigated in the present study. We had already reported30 an interaction between T. versicolor laccase (PDB ID: 1GYC) and the phenolic substrates by molecular docking simulations.
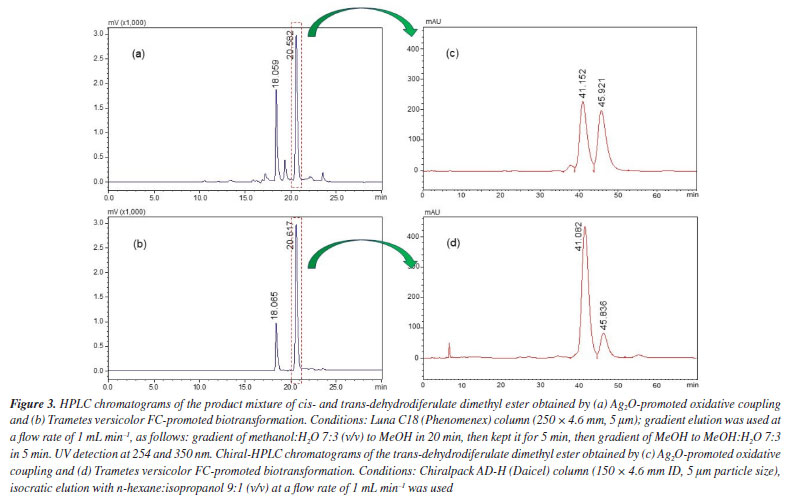
The biotransformation was scaled up to purify and characterize the main biotransformation product unequivocally. NMR analyses (see Supplementary Material, Figures 1S-3S) of the biotransformation product revealed that it had the same chemical structure as DBN 2. Previously, we reported the complete and unequivocal assignments of 1H and 13C NMR data for DBN 2, leaving no ambiguities and including the measurement of all hydrogen homonuclear coupling constant values. Density functional theory (DFT) calculations, nuclear Overhauser effect (NOE) experiments, and J values measurements confirmed the relative stereochemistry. All DBN 2 1H and 13C data agreed with the data previously reported32 for trans-dehydrodiferulate dimethyl ester. Finally, the stereoselectivity of the biotransformation approach developed herein was determined by chiral HPLC analysis. Comparison between the C18-HPLC chromatograms of the major biotransformation product and a sample of synthetic DBN 2 previously available in our laboratory revealed similar profiles, with peaks at 18.0 and 20.6 min corresponding to dimethyl cis- and trans-dehydrodiferulate dimethyl ester (2), respectively (Figures 3a and 3b). Next, the major peaks corresponding to the trans-enantiomers of both biotransformation and synthetic products were isolated and analyzed by chiral HPLC. The chromatogram of trans-DBN 2 obtained by the synthetic approach (i.e., Ag2O-promoted oxidative coupling of 1) displayed two peaks (Figure 3c), at 41.15 and 45.92 min, with similar areas (48.1 and 48.9%, respectively). This indicated the presence of the two trans-enantiomers at approximately the same concentrations and a lack of enantioselectivity. On the other hand, the chromatogram of the biotransformation product (Figure 3d) displayed two peaks, at the same retention times mentioned above, with different relative areas (81.7 and 16.3%, respectively). This pointed out that one of the enantiomers of DBN 2 was preferentially formed with an enantiomeric excess of around 66.7% of the main enantiomer. Although no further experiment was carried out to identify the most abundant enantiomer in the mixture of biotransformation products, these preliminary results indicated that the biotransformation approach led to an enantiomeric excess that could not be achieved by the synthetic approach.
The specialized literature contains reports on approaches similar to the one we developed herein, aiming at the coupling of phenylpropanoid units. The total synthesis of the β-5/β-O-4 dihydroxytrimer and dihydrotrimer of coniferyl alcohol were obtained in global yield, 9 and 20%, respectively, over nine steps starting from ferulic acid using laccase-mediated dimerization, nucleophilic substitution, and dihydroxylation.46 However, the study did not show the enantioselectivity of the reaction. Despite some limitations, which are mainly correlated to the toxicity of some substrates and products to the fungal cell, which could limit the amount of starting materials in biotransformation reactions, our approach is promising. Our results showed good yield (higher than most literature reports), diastereo-, and enantioselectivity, along with fewer reactional steps.
CONCLUSIONS The soil fungal strain (T. versicolor FC) was screened as a potential biocatalyst that can efficiently and selectively biotransform methyl trans-ferulate (1) into the corresponding dihydrobenzofuran neolignan 2 ((±)-trans-dehydrodiferulate dimethyl ester). Our approach produced DBN 2 with good enantioselectivity, under mild reaction conditions, using nontoxic and inexpensive reagents, aligning with the principles of green catalysis. These preliminary results indicate that combining biotransformation and other chemical processes may be an excellent starting point to obtain dihydrobenzofuran neolignans from the corresponding phenylpropanoids enantioselectively as an alternative to traditional synthetic methods. It is worth mentioning that further optimization studies will be necessary to reach higher enantiomeric excess (close to 100%), which is mandatory for new drug development programs.
SUPPLEMENTARY MATERIAL 1H and 13C NMR, and DEPT 135 spectra, and high-resolution mass spectra of compound 2 are available at http://quimicanova.sbq.org.br, as a PDF file, free of charge.
DATA AVAILABILITY STATEMENT All data are available in the text and supplementary material.
ACKNOWLEDGMENTS The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant number 2013/20094-0 and 2021/10098-4) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, proc. 310648/2022-0 and 306186/2021-7) for financial support, and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES, finance code 001) for fellowships.
REFERENCES 1. Moss, G. P.; Pure Appl. Chem. 2000, 72, 1493. [Crossref] 2. Pagotti, M. C.; Dias, H. J.; Candido, A. C. B. B.; Oliveira, T. A. S.; Borges, A.; Oliveira, N. D.; Lopes, C. D.; Orenha, R. P.; Parreira, R. L. T.; Crotti, A. E. M.; Magalhães, L. G.; Pharmaceutics 2023, 15, 754. [Crossref] 3. Baldin, E. L. L.; Dias, H. J.; de Souza, C. M.; Soares, M. C. E.; Grundman, C. O.; Santos, T. L. B.; Crotti, A. E. M.; J. Pest Sci. 2019, 92, 861. [Crossref] 4. Dias, H. J.; Patrocínio, A. B.; Pagotti, M. C.; Fukui, M. J.; Rodrigues, V.; Magalhães, L. G.; Crotti, A. E. M.; Chem. Biodiversity 2018, 15, e1800134. [Crossref] 5. Oliveira, L. G. C.; Brito, L. M.; Alves, M. M. M.; Amorim, L. V.; Sobrinho-Júnior, E. P. C.; de Carvalho, C. E. S.; Rodrigues, K. A. F.; Arcanjo, D. D. R.; Citó, A. M. G. L.; Carvalho, F. A. A.; Basic Clin. Pharmacol. Toxicol. 2017, 120, 52. [Crossref] 6. Farhat, J.; Alzyoud, L.; Alwahsh, M.; Al-Omari, B.; Cancers 2022, 14, 2196. [Crossref] 7. Pieters, L.; Van Dyck, S.; Gao, M.; Bai, R.; Hamel, E.; Vlietinck, A.; Lemière, G.; J. Med. Chem. 1999, 42, 5475. [Crossref] 8. Ma, S.-J.; Li, H.-B.; Li, T.; Su, Z.-Z.; Wang, Z.-Z.; Yao, X.-S.; Xiao, W.; Yu, Y.; RSC Adv. 2021, 11, 30725. [Crossref] 9. Huang, X.-X.; Zhou, C.-C.; Li, L.-Z.; Peng, Y.; Lou, L.-L.; Liu, S.; Li, D.-M.; Ikejima, T.; Song, S.-J.; Fitoterapia 2013, 91, 217. [Crossref] 10. Dias, H. J.; Rodrigues, M. L.; Crotti, A. E. M.; J. Braz. Chem. Soc. 2021, 32, 20. [Crossref] 11. Silva, A. R.; Polo, E. C.; Martins, N. C.; Correia, C. R. D.; Adv. Synth. Catal. 2018, 360, 346. [Crossref] 12. Dias, H. J.; Silva, E. O.; Vieira, T. M.; Crotti, A. E. M. In Benzofuran: Production and Applications; Barros, M. S., ed.; Nova Science Publishers: New York, 2020, ch. 2. 13. Daquino, C.; Rescifina, A.; Spatafora, C.; Tringali, C.; Eur. J. Org. Chem. 2009, 2009, 6289. [Crossref] 14. Kuo, Y.-H.; Wu, C.-H.; J. Nat. Prod. 1996, 59, 625. [Crossref] 15. Blum, T. R.; Zhu, Y.; Nordeen, S. A.; Yoon, T. P.; Angew. Chem. 2014, 126, 11236. [Crossref] 16. Lemière, G.; Gao, M.; De Groot, A.; Dommisse, R.; Lepoivre, J.; Pieters, L.; Buss, V.; J. Chem. Soc., Perkin Trans. 1 1995, 1775. [Crossref] 17. Gupta, D.; Bleakley, B.; Gupta, R. K.; J. Ethnopharmacol. 2008, 115, 361. [Crossref] 18. Majhi, S.; Curr. Org. Chem. 2021, 25, 1047. [Crossref] 19. Majhi, S.; Ultrason. Sonochem. 2021, 77, 105665. [Crossref] 20. Li, J.; Amatuni, A.; Renata, H.; Curr. Opin. Chem. Biol. 2020, 55, 111. [Crossref] 21. dos Santos, V. H. P.; Coelho Neto, D. M.; Lacerda Júnior, V.; Borges, W. S.; Silva, E. O.; Curr. Org. Chem. 2020, 24, 2902. [Crossref] 22. Saliu, F.; Tolppa, E.-L.; Zoia, L.; Orlandi, M.; Tetrahedron Lett. 2011, 52, 3856. [Crossref] 23. Nassiri-Koopaei, N.; Faramarzi, M. A.; Biocatal. Biotransform. 2015, 33, 1. [Crossref] 24. Hawksworth, D. L.; Mycol. Res. 2001, 105, 1422. [Crossref] 25. Kuhar, F.; Castiglia, V.; Levin, L.; Int. Biodeterior. Biodegrad. 2015, 104, 238. [Crossref] 26. Liers, C.; Pecyna, M. J.; Kellner, H.; Worrich, A.; Zorn, H.; Steffen, K. T.; Hofrichter, M.; Ullrich, R.; Appl. Microbiol. Biotechnol. 2013, 97, 5839. [Crossref] 27. Giardina, P.; Faraco, V.; Pezzella, C.; Piscitelli, A.; Vanhulle, S.; Sannia, G.; Cell. Mol. Life Sci. 2010, 67, 369. [Crossref] 28. Riva, S.; Trends Biotechnol. 2006, 24, 219. [Crossref] 29. Pinto, P. A.; Fraga, I.; Bezerra, R. M. F.; Dias, A. A.; Biochem. Eng. J. 2020, 153, 107423. [Crossref] 30. Conceição, J. C. S.; Dias, H. J.; Peralva, C. M. S.; Crotti, A. E. M.; Pita, S. S. R.; Silva, E. O.; Appl. Biochem. Biotechnol. 2020, 190, 1498. [Crossref] 31. Fukui, M. J.; Dias, H. J.; Severiano, M. E.; de Souza, M. G. M.; de Oliveira, P. F.; Ambrósio, S. R.; Martins, C. H. G.; Tavares, D. C.; Crotti, A. E. M.; ChemistrySelect 2018, 3, 1836. [Crossref] 32. Medeiros, T. C. T.; Dias, H. J.; Silva, E. O.; Fukui, M. J.; Soares, A. C. F.; Kar, T.; Heleno, V. C. G.; Donate, P. M.; Parreira, R. L. T.; Crotti, A. E. M.; J. Braz. Chem. Soc. 2016, 27, 136. [Crossref] 33. Aamir, S.; Sutar, S.; Singh, S. K.; Baghela, A.; Plant Pathology & Quarantine 2015, 5, 74. [Crossref] 34. Hall, T. A.; Nucleic Acids Symp. Ser. 1999, 41, 95. [Link] accessed in May 2025 35. Thompson, J. D.; Gibson, T. J.; Plewniak, F.; Jeanmougin, F.; Higgins, D. G.; Nucleic Acids Res. 1997, 25, 4876. [Crossref] 36. Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S.; Mol. Biol. Evol. 2013, 30, 2725. [Crossref] 37. Saitou, N.; Nei, M.; Mol. Biol. Evol. 1987, 4, 406. [Crossref] 38. Kimura, M.; J. Mol. Evol. 1980, 16, 111. [Crossref] 39. Felsenstein, J.; Evolution 1985, 39, 783. [Crossref] 40. Niku-Paavola, M. L.; Raaska, L.; Itävaara, M.; Mycol. Res. 1990, 94, 27. [Crossref] 41. Wang, F.; Yu, X.; Yu, Z.; Cui, Y.; Xu, L.; Huo, S.; Ding, Z.; Zhao, L.; Du, L.; Qiu, Y.; Front. Bioeng. Biotechnol. 2023, 11, 1176352. [Crossref] 42. Akpinar, M.; Urek, R. O.; 3 Biotech 2017, 7, 98. [Crossref] 43. Raja, H. A.; Miller, A. N.; Pearce, C. J.; Oberlies, N. H.; J. Nat. Prod. 2017, 80, 756. [Crossref] 44. Martínková, L.; Kotik, M.; Marková, E.; Homolka, L.; Chemosphere 2016, 149, 373. [Crossref] 45. Atilano-Camino, M. M.; Álvarez-Valencia, L. H.; García-González, A.; García-Reyes, R. B.; J. Environ. Manage. 2020, 275, 111231. [Crossref] 46. Flourat, A. L.; Peru, A. A. M.; Haudrechy, A.; Renault, J.-H.; Allais, F.; Front. Chem. 2019, 7, 842. [Crossref]
Guest Editor handled this article: Lucas S. Abreu |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access