
 |
 |
 |
 |
 |
Artigo
| Regioselective and chemoselective biocatalytic reduction of α,β,γ,δ-unsaturated ketones by marine-derived fungus Penicillium citrinum CBMAI 1186 |
|
Pedro H. DamadaI,* I. Laboratório de Química Orgânica e Biocatálise, Instituto de Química de São Carlos, Universidade de São Paulo, 13563-120 São Carlos - SP, Brasil Received: 03/13/2025 *e-mail: pedrohdamada@gmail.com Fungi exhibits remarkable adaptability to diverse environments, making them widely distributed across ecosystems. Their vast diversity has driven extensive research into biotechnological applications, including biocatalysis mediated by whole cells or isolated enzymes. Among fungal biocatalysts, marine-derived strains have gained attention due to their unique enzymatic repertoire. In this study, whole cells of Penicillium citrinum CBMAI 1186 were employed to catalyze the selective reduction of α, β, γ, δ-unsaturated ketones into γ, δ-unsaturated ketones. The substrates were synthesized and characterized by nuclear magnetic resonance (NMR) and gas chromatography-mass spectrometry (GC-MS), while crystalline forms were further analyzed via X-ray diffraction. P. citrinum CBMAI 1186 exhibited high catalytic efficiency, achieving conversions exceeding 85% after six days. Fluorine- and methyl-substituted compounds displayed faster conversion (1-3 days), suggesting substrate preference. In all cases, only the α, β-double bond was reduced, confirming the selectivity of the microorganism for highly conjugated compounds. These discoveries indicate that P. citrinum CBMAI 1186 performs regioselective and chemoselective reductions with high efficiency, establishing its potential as a biocatalyst for selective bioreduction reactions. The exclusive transformation of α, β-unsaturated bonds underscores its applicability in biotechnological processes. Overall, this study highlights the potential of P. citrinum CBMAI 1186 in the development of novel sustainable bioprocesses for fine chemical synthesis. INTRODUCTION Biocatalysts have emerged as superior alternatives to conventional chemical catalysts due to their capacity to generate high-value products across various industries, including food, pharmaceuticals, and fine chemistry, while conducting cost-effective reactions with reduced environmental impact.1,2 Enzymes, the most employed biocatalysts, can be utilized either in their isolated form or within whole cells, such as bacteria and fungi. Although isolated enzymes provide accurate control over reactions and demonstrate high efficiency, whole-cell biocatalysts typically display resilience due to the protective barrier they receive against environmental stressors such as aeration and reactive substrates or products. This setup provides an optimal environment for enzymes, ensuring structural stability even in challenging conditions and facilitating efficient cofactor regeneration while effectively orchestrating reaction cascades.3,4 Moreover, the utilization of whole cells from microorganisms is promising because of the plethora of novel enzymes capable of synthesizing compounds of considerable interest.5-7 Over the course of evolution, microorganisms have encountered various substrates and environmental conditions, resulting in the development of numerous metabolites and enzymatic activities.3 While bacteria are more frequently employed than fungi in biocatalytic reactions due to their ease of application, fungi have attracted attention in recent years for their remarkable adaptability under laboratory conditions and the greater diversity of species they offer.8-10 In this study, we investigate the utilization of whole cells from Penicillium citrinum CBMAI 1186 to catalyze the conversion of α, β, γ, δ-unsaturated ketones into γ, δ-unsaturated ketones. It is noteworthy that existing literature primarily emphasizes α, β-unsaturated compounds, while α, β, γ, δ-unsaturated compounds have received comparatively limited attention. This highlights the significance of acquiring additional data to enhance comprehension of the reaction conditions and broaden the scope of substrates for these reactions. In our group, a similar reaction was prepared using an α,β,γ,δ-unsaturated ketone. In this study, we expanded the range of substrate to confirm fungal activity, tested different reaction times, and determined whether previously tested conditions, like a biphasic system, are necessary for high-yield reactions. The reduction of α, β-unsaturated compounds is extensively investigated and stands as a pivotal tool in organic synthesis, chiefly due to its ability to generate chiral centers, resulting in compounds with high enantiomeric/stereoselectivity purity - products of notable interest.11,12 Conversely, the reduction of α, β, γ, δ-unsaturated carbonyl compounds presents significant challenges owing to the presence of three reducible positions. The complexity arises from the existence of these multiple reducible positions (e.g., 1,2-, 1,4-, and 1,6-reduction), potentially yielding mixtures of multi-reduced products under reductive conditions. Since γ, δ-unsaturated alkenes are vital for the synthesis of important chemicals, the discovery of an effective catalyst for selectively reduce α, β, γ, δ-unsaturated alkenes into γ, δ-unsaturated alkenes in an environmentally friendly manner holds utmost importance.13,14
EXPERIMENTAL Chemical reagents, solvents, and culture media The compounds trans-cinnamaldehyde (1) (98%) and 4-iodoacetophenone (2d) were purchased from Oakwood Chemical® (USA). 4-Bromoacetophenone (2a) (98%), 4-fluoroacetophenone (2c) (99%), 4-nitroacetophenone (2e) (98%), and 4-methylacetophenone (2f) (95%), were purchased from Sigma-Aldrich® (USA). 4-Chloro-acetophenone (2b) (98%) was purchased from Acros Organics® (Belgium). Sodium hydroxide (97%) was purchased from Quimis (Brazil). Deuterated chloroform (CDCl3) (> 99%) was purchased from Cambridge Isotope Laboratories (USA). The solvents acetone, hexane, ethyl acetate and dimethyl sulfoxide (DMSO) were purchased from Exodo, Merck and Synth (Brazil). All compounds were used without further purification. The synthetized compounds were purified by column chromatography using flash silica gel with pore diameter of 6 nm (0.035-0.070 nm) which was obtained from Acros Organics®. Thin layer chromatography (TLC) (Macherey-Nagel, 20 × 20 cm, silica gel 60, G / UV 254 nm) was used with the purpose of monitor the synthesis reactions. The salts used for preparation of the synthetic sea water (see "Preparation of culture medium and synthetic sea water" sub-section) were purchased from Synth and Vetec (Brazil). Agar and malt extract were purchased from Kasvi (Brazil). Preparation of the α,β,γ,δ-unsaturated ketones (3a-3f) The α,β,γ,δ-unsaturated ketones were synthesized through Claisen-Schmidt condensation. To obtain the products (3a-3f), trans-cinnamaldehyde (1) (3.1 mmol, excess) and the corresponding ketones (2a-2f, 3 mmol) were used. All the reactants were transferred into a 25 mL rounded-bottomed flask containing 10-15 mL of ethanol. Next, 0.3 mL of 6 M aqueous sodium hydroxide solution (NaOH) was slowly added. The reactions were agitated at ambient temperature for 0.5 h and monitored via thin layer chromatography (TLC). The resulting precipitates were isolated using Buchner filtration and rinsed with chilled distilled water until reaching a neutral pH. The precipitates were then dried under room temperature (RT) and washed 3 times with boiling hexane. The products (4a-4f) were obtained in good yields and were identified and characterized by 1H and 13C nuclear magnetic resonance (NMR), Fourier-transform infrared spectroscopy (FTIR), gas chromatography-mass spectrometry (GC-MS), and melting point (mp) analyses (see Supplementary Material). Single crystal X-ray diffraction analyses The compounds 3c, 3d and 3f were obtained as crystals and submitted to X-ray diffraction. The X-ray data collection for 3c, 3d and 3f were performed at 100 K, using the MANACÁ beamline of the Brazilian National Synchrotron Light Laboratory (LNLS, Campinas, Brazil), equipped with a Pilatus M2 detector and an MK3 mini-kappa, using phi-scans with 360º rotation in steps of 0.3º, and X-ray wavelength regulated to 0.67019 Å. The XDS software15 was employed for unit cell refinement, data collection and reduction, and empirical absorption correction. The structures were solved with the SHELXT program, using the Intrinsic Phasing method,16 while the non-hydrogen atoms were refined by the least-squares minimization method using the SHELXL program17 and considering anisotropic displacement parameters, both within the Olex2 program.18 Hydrogen atoms were positioned at calculated positions and refined with the riding model. Olex 2 and Mercury 2022.2.019 were used to prepare the graphical illustrations. Data collection and refinement parameters are listed in Supplementary Material (Table 1S). Crystallographic data of 3c, 3d and 3f in CIF format have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the deposition numbers CCDC 2283823, CCDC 2283824 and CCDC 2283825, respectively. Copies of the data can be obtained free of charge from the CCDC website (www.ccdc.cam.ac.uk). Marine-derived fungus P. citrinum CBMAI 1186 Marine-derived fungus P. citrinum CBMAI 1186 was isolated from the marine alga Caulerpa sp. collected by Prof. R. G. S. Berlinck in the city of São Sebastião, on the coast of the state of São Paulo, Brazil. The fungus was identified using conventional and molecular methods at the Chemical, Biological and Agricultural Pluridisciplinary Research Center (CPQBA) at the University of Campinas (Brazil). A type of culture of P. citrinum CBMAI 1186 was deposited in the Brazilian Collection of Environmental and Industrial Microorganisms (CBMAI) located at CPQBA/UNICAMP (https://cbmai.cpqba.unicamp.br/) a microbial culture collection that serves as a repository for microorganisms isolated from various environments, including soil, water, and industrial processes. The collection is maintained to preserve microbial biodiversity and provide strains for scientific research and biotechnological applications. Preparation of culture medium and synthetic sea water To cultivate the fungus P. citrinum CBMAI 1186, both solid and liquid culture media were employed. The composition of both media was identical, consisting of malt extract (20 g L-1) dissolved in synthetic sea water with a pH 8. The only difference between the two media was that the solid media included agar (20 g L-1) as a solidifying agent. The synthetic sea water was prepared with the following salts: CaCl2.2H2O (1.36 g L-1), MgCl2.6H2O (9.68 g L-1), KCl (0.61 g L-1), NaCl (30.0 g L-1), Na2HPO4 (0.014 mg L-1), Na2SO4 (3.47 g L-1), NaHCO3 (0.17 g L-1), KBr (0.1 g L-1), SrCl2.6H2O (0.040 g L-1), and H3BO3 (0.030 g L-1). All manipulations involving the marine-derived fungus P. citrinum CBMAI 1186 were carried out under sterile conditions in a Veco laminar flow hood. Growth of marine-derived fungus P. citrinum CBMAI 1186 in solid and liquid culture media From a stock medium stored at 4 ºC, the fungus P. citrinum CBMAI 1186 was transferred into a Petri plate containing fresh solid culture media. The fungus was cultured under optimized conditions. Following 7 days of incubation at 32 ºC, 10-15 slices (0.3 cm × 0.3 cm) containing fungal mycelia were removed and transferred to Erlenmeyer flasks (250 mL) with 100 mL of synthetic seawater and malt extract. The cultures were then maintained at 32 ºC. After 7 days of incubation on an orbital shaker (130 rpm, 32 ºC), the mycelia were filtered via Buchner funnel apparatus. The mycelia were used for biocatalytic reduction of α,β,γ,δ-unsaturated ketones 3a-3f. Biocatalytic reduction of α,β,γ,δ-unsaturated ketones 3a-3f from P. citrinum CBMAI 1186 The mycelia of the fungus P. citrinum CBMAI 1186 were harvested by Buchner filtration and suspended in 25 mL of phosphate buffer solution (Na2HPO4/KH2PO4, pH = 7.1, 0.1 mol L-1) added in a 125 mL-Erlenmeyer flask. The biocatalytic reductions were carried out with 2.5 g (wet weight) of mycelium and 25 mg of α,β-unsaturated ketones 3a-3f, previously dissolved in DMSO (0.4-1 mL). The mixtures were incubated in an orbital shaker for 1, 3 and 6 days, at 32 ºC and 130 rpm. After each period determined, the reactions were stopped by adding 50 mL of ethyl acetate (EtOAc). In order to obtain the product, the mycelia, in EtOAc, was submitted to vigorous agitation for 30 min. The reactions for 1 and 3 days were not repeated, and for 6 days was made in duplicate. Isolation of products 4a-4f from biocatalytic reduction by P. citrinum CBMAI 1186 After the end of the biocatalytic reaction, the mycelia were isolated from the reaction mixture via filtration using a Buchner funnel, after which they were discarded. To enhance product yield, the extraction with ethyl acetate was conducted thrice, employing the following procedure for each repetition: 30 mL of EtOAc was introduced into the aqueous filtrate within a 125 mL Erlenmeyer flask and vigorously agitated using a magnetic stirrer. Subsequently, the resultant mixture was transferred to a separation funnel, where the organic phase was separated from the aqueous phase. The organic phase obtained from each extraction was collected in a flask, desiccated over anhydrous sodium sulfate (Na2SO4), filtered, subjected to vacuum evaporation, and analyzed using GC-MS. The compounds were purified through flash column chromatography (CC) utilizing silica gel, resulting in the isolation of the purified products 4a-4f, whose purity was confirmed by TLC. Products 4a-4f underwent identification and characterization via NMR, GC-MS, and FTIR analyses. Analytical methods For GC-MS, a Shimadzu GC2010 Plus gas chromatography system coupled to a mass-selective detector (MS) (Shimadzu MS2010 Plus, Kyoto, Japan) in electron ionization mode (70 eV) was used. The samples were injected into a DB5 column (30 m × 0.25 mm × 0.25 μm, J&W Scientific, Folsom, CA), and the following conditions were applied: the temperatures of the injector and interface were maintained at 250 and 270 ºC, respectively. While, for the oven, the initial temperature was 90 ºC, and was held for 4 min, after this period, the temperature was increased at 10 ºC min-1 until 280 ºC, which was held for 5 min, and then, at 10 ºC min-1, the temperature was increased to 300 ºC and held for 10 min. The total analysis time was 40 min. Helium was used as carrier gas with initial flow of 0.75 mL min-1, and the injection volume was 1 μL (split ratio of 1:20). Fragment ions were detected in the range of m/z 40-550. The FTIR spectra were recorded on a Bruker (Alpha model) spectrometer in the 4000-400 cm-1 region. The samples were first solubilized in dichloromethane and placed in the analysis area provided with an infrared window made of zinc selenide (ZnSe) crystal. 1H and 13C NMR spectra were recorded on an Agilent Technologies 400/54 Premium Shielded spectrometer, operating at 400 MHz for 1H and 100 MHz for 13C. The chemical shifts (d) were given in parts per million (ppm) and coupling constants (J) in Hz. For the analysis, deuterated chloroform (CDCl3) was used to solubilize the compounds (dH = 7.25 ppm, s; δC = 77.00, t) and tetramethysilane (TMS) as an internal standard. Melting points were measured on a Fisatom (model 431) by observing the phase changing of the samples.
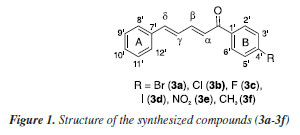
RESULTS AND DISCUSSION Synthesis of α,β,γ,δ-unsaturated ketones 3a-3f and characterization of the (2E,4E)-1-(4-fluorophenyl)-5-phenylpenta-2,4-dien-1-one (3c) The α,β,γ,δ-unsaturated ketones 3a-3f (Figure 1) were synthesized using the Claisen-Schmidt condensation reaction, yielding satisfactory results ranging from 65 to 85%, indicating that the reaction conditions were suitable.20-22 All synthesized compounds were obtained as yellow solids. The presence of different substituents on the B-ring did not appear to have any significant correlation with the reaction yields.
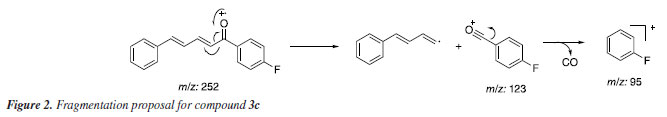
The Claisen-Schmidt reaction is an aldol condensation between an aromatic aldehyde and a carbonyl compound, catalyzed by a base, resulting in an α,β-unsaturated carbonyl compound, typically named as chalcone. The base generates an enolate ion from the carbonyl compound, which then attacks the aldehyde, followed by dehydration (condensation) to form the desired product. The compounds 3a-3f were identified by 1H and 13C NMR, FTIR, and GC-MS analyses, and their corresponding spectra are available in the Supplementary Material. Considering their structural resemblances, compound 3c was selected as a representative model for comprehensive characterization analyses. Within the mass spectrum of compound 3c, two peaks of significant intensity were observed. The first peak appeared at m/z 252 (100%) value, corresponding to the molecular ion [C17H13FO]+. The second peak was detected at m/z 123 (35%), indicating the presence of an acylium ion [C7H4FO]+, formed through fragmentation processes. A peak at m/z 95 indicates the potential loss of a CO molecule from the acylium ion. A proposed fragmentation of compound 3c is presented in Figure 2.
For the 1H NMR spectrum (400 MHz, CDCl3), the hydrogens adjacent to the double bonds exhibited multiple signals due to the influence of both electronic effects and vicinal coupling, leading to characteristic splitting patterns. Notably, the Hα signal appeared at 7.07 ppm as a doublet with a coupling constant (J) of 15 Hz, which is consistent with a trans-alkene. The magnitude of this coupling aligns with literature23 values for alkenic protons in an E-configuration, confirming the geometric arrangement of the double bond. In the 13C NMR spectrum (100 MHz, CDCl3), the most unshielded signals were observed at 188.9 ppm, corresponding to the carbonyl carbon (C=O), and at 165 ppm, attributed to the carbon directly bonded to fluorine group. Atoms with high electronegativity withdraw the electron density from the adjacent carbon, shifting its resonance significantly downfield. Additionally, the large one-bond carbon-fluorine coupling constant (J1C-F = 254 Hz) confirms the direct C-F bond and further supports the assignment of this signal. The large coupling constant, typically in the range of 200-300 Hz, is attributed to the high electronegativity of fluorine, strong C-F bond polarization, and efficient orbital overlap between carbon and fluorine. This phenomenon is a key spectral feature of organofluorine compounds and provides valuable structural and electronic information.23 The appearance of two peaks of equal intensity for the carbon bonded to fluorine in the 13C NMR spectrum is due to the direct one-bond coupling (J1C-F) with 19F, resulting in a characteristic doublet.23 A total of 13 distinct carbon signals were observed in the 13C NMR spectrum, corresponding to 17 carbon atoms in the molecular structure. This discrepancy suggests the presence of symmetrical elements in the molecule, which leads to overlapping signals for chemically equivalent carbons. The observation that all carbons exhibit chemical shifts typical of sp2-type hybridized carbons further supports the presence of a fully conjugated system, reinforcing the structural assignment (Figure 31S in the Supplementary Material). Structural crystal description of 3c, 3d and 3f The compounds 3c, 3d, and 3f were obtained in crystalline form and crystals from each compound were isolated for analysis using the single crystal X-ray diffraction technique. The findings, including ORTEP illustrations and selected bond lengths for the three compounds, were detailed in the Supplementary Material (Figure 1S, Tables 2S and 3S). The X-ray diffraction analysis revealed that compounds 3c and 3d crystallized in the orthorhombic space groups P212121 and Pca21, respectively. Conversely, compound 3f exhibited crystallization in the monoclinic space group P21/c. Among these compounds, 3d and 3f displayed one molecule per asymmetric unit, whereas the crystal structure of compound 3c contained two distinct molecules per asymmetric unit denoted as 3c' and 3c". Notably, these molecules exhibited differentiation in certain torsion angles, such as those observed for the carbon atoms C6-C7-C8-C9, which displayed torsion angles of -164.12º and 161.74º in 3c' and 3c", respectively. As anticipated for a conjugated system, the bond angles within the α,β,γ,δ-unsaturated ketone skeleton closely approximate 120º. In the solid-state structure, compounds 3d and 3f exhibited nearly planar configurations, with dihedral angles between the aromatic groups measuring 7.47º and 10.17º, respectively. However, compound 3c deviates from planarity, displaying dihedral angles of 44.21º and 43.15º (Figure 2S, Supplementary Material). Reduction of α,β,γ,δ-unsaturated ketones 3a-3f by whole cells of P. citrinum CBMAI 1186 Biocatalytic reactions using whole cells of P. citrinum CBMΑI 1186 were carried out on α,β,γ,δ-unsaturated ketones 3a-3f, previously characterized, for periods of 1, 3, and 6 days. After 6 days, the conversion rates for all compounds exceeded 85%, as determined by analyzing the respective areas in the GC-MS chromatograms (Table 1). It is worth noting that prior studies24,25 have investigated the reduction of double bonds using other fungi, primarily focusing on α,β-unsaturated compounds.
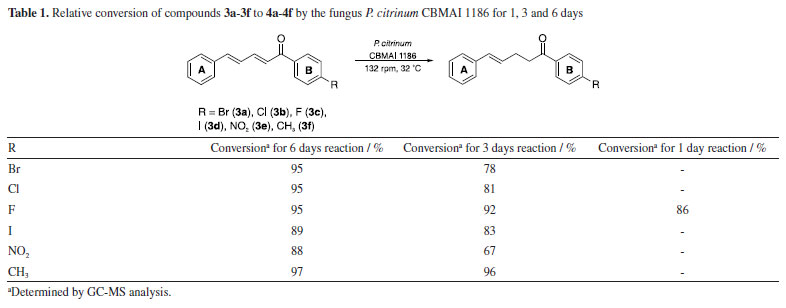
Regarding the 3-day reaction, conversion values exceeding 90% were observed for compounds 3c and 3f, suggesting an elevated affinity of these substrates for the fungal cells and its enzymes. Compound 3c included a fluorine atom in the para-position, acting as an electron-withdrawing group, while compound 3f contained a methyl group in the para-position, acting as an electron-donating group. The distinct electronic effects of these groups on the aromatic ring might impact the reaction by altering electronic density and potentially influencing enzyme activity towards the double bond. However, it was observed that the presence of these groups did not affect the reaction conditions. The observed outcome can be attributed to the minimal steric effects afforded by the small size of the fluorine atom and the methyl group, which facilitated efficient biotransformation. Consequently, this resulted in a reduced reaction time compared to other compounds (3a, 3b, 3d, and 3e). The conversion achieved after a 3-day reaction was significant, given that many biotransformations typically require more than five days to reach completion,26,27 indicating favorable acceptance of the compound by cells. This observation was further supported by the results of a 1-day reaction involving compound 3c, which yielded an 86% conversion rate in the production of compound 4c (Table 1). The rapid reaction times observed reinforce the importance of understanding the substrate structure and the relation with the cells for optimizing results, as it can directly influence efficiency and lead to higher conversion rates in shorter periods. In a previous study conducted by our research group,28 the compounds 3c and 3f were used as substrates in biocatalytic reactions catalyzed by ene-reductases (ER) from Penicillium steckii, a fungus of the same genus. For these reactions, Escherichia coli cells were transformed with the target genes, and whole-cell biotransformation were performed. The products were identical to those obtained in our present study, suggesting the involvement of a similar enzymatic system. Ene-reductases are oxirreductases that catalyze the reduction of C=C double bonds in α,β-unsaturated compounds, where the double bond is conjugated to an electron-withdrawing group (EWG), such as a carbonyl moiety. These enzymes have been extensively studied since the first member, OYE1 (old yellow enzyme), was identified in 1932 as the first flavin-dependent enzyme. Since then, various ER families have been described, with the OYE family remaining the most well-characterized. Within this family, different classes have been defined, with classes I and II being the most extensively studied.29,30 The catalytic mechanism consists of two sequential half-reactions. In the reductive half-reaction, a hydride ion is transferred from NAD(P)H to the enzyme-bound flavin cofactor (FMN), reducing it. In the oxidative half-reaction, the reduced flavin transfers a hydride to the β-carbon of the substrate, adjacent to the EWG, while a proton from a conserved tyrosine residue is transferred to the α-carbon. This reaction follows a ping-pong mechanism, leading to a trans-hydrogen atom transfer process capable of generating chiral centers depending on the structures of the conjugated compounds. The enantioselectivity is a key feature that makes ERs valuable biocatalysts for the synthesis of enantiopure compounds.11,31 In contrast to our current approach, in the reaction with E. coli and the target genes, hexane was added to the reaction medium to facilitate the process, the same procedure implemented by Ferreira et al.32 to enhance the reduction efficiency. The conversion rates of two of the tested enzymes, PsOYE3 and PsOYE4, exceeded 80% after 24 h. Notably, in our study, the same substrate was reduced to a comparable extent within 24 h without the need for hexane, demonstrating a better substrate acceptance by fungal cells compared to E. coli. This improved performance may be attributed to differences in the composition and permeability of the membrane, which could enhance substrate uptake and product release, thereby facilitating a more efficient biotransformation process. Another indication of an ene-reductase is the reduction of only one double bond, specifically the one near the EWG group (as discussed in the next section). This highlights the need for additional genomic and biochemical analyses of P. citrinum CBMAI 1186 to accurately understand its biocatalytic activity. Whole cell biocatalysis has proven to be a versatile and efficient approach, offering a robust, cost-effective system that requires minimal preparation while enabling high product yields. However, the use of whole cells also presents challenges. Although the enzyme responsible for the observed activity can catalyze the formation of stereoisomers, its presence inside the cell introduces challenges, as other intracellular enzymes may promote side reactions that reduce stereoselectivity. Within the cellular environment, multiple enzymes can act simultaneously, which can lead to competing transformations that alter the desired product profile. One key example that could occur in our study is the unintended reduction of carbonyl groups, which may result in the formation of alcohols. To prevent the reduction of the carbonyl group, this study employed a strategic approach by introducing a bulky aromatic ring near the carbonyl, creating steric hindrance. This effectively preserved the carbonyl functionality during the reduction process catalyzed by P. citrinum CBMAI 1186. This strategy is particularly relevant due to the presence of intracellular enzymes, such as alcohol dehydrogenases,33 which readily reduce carbonyl groups to alcohols. By carefully designing substrate structures or optimizing reaction conditions, side reactions can be minimized, enhancing the selectivity of whole cell biocatalysis. The addition of a bulky group was inspired by a study by Ferreira et al.,34 in which P. citrinum CBMAI 1186 mycelia were used for the biotransformation of (E)-2-methyl-3-phenylacrylaldehyde. The primary product, 2-methyl-3-phenylpropan-1-ol, suggested that the carbonyl group underwent reduction alongside the α,β-bond. Isolation of the products 4a-4f obtained by biocatalytic reduction of compounds 3a-3f using P. citrinum CBMAI 1186 The products 4a-4f (Table 2) resulting from the 6-day biocatalytic reactions of compounds 3a-3f utilizing P. citrinum CBMAI 1186 were isolated and purified via column chromatography. Following purification, these products 4a-4f underwent identification and characterization through FTIR, GC-MS, and 1H and 13C NMR analyses. All relevant spectra and chromatograms were provided in the Supplementary Material.
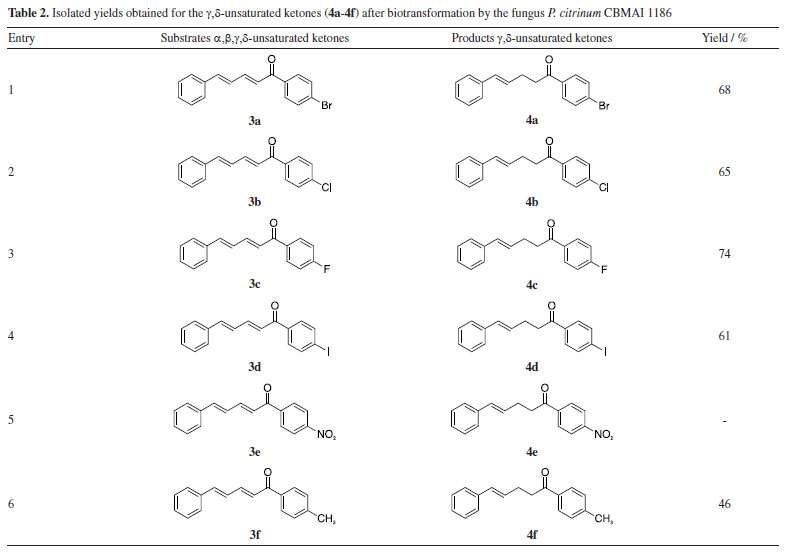
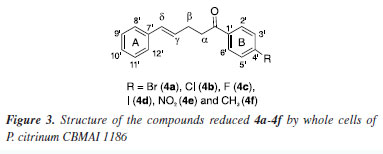
During the purification process employing hexane and either ethyl acetate or chloroform, the separation of compound 4e from substrate 3e proved challenging due to its persistence. The yields obtained after purifying products 4a-4d and 4f via column chromatography are presented in Table 2. The products were obtained in oil form, in contrast to the solid format of the initial compounds. The confirmation of α,β-bond reduction in substrates 3a-3f was accomplished through analytical techniques. GC-MS analysis revealed an increase of 2 atomic mass units (amu) in the molecular mass of compounds 4a-4d and 4f, indicating the incorporation of two hydrogen atoms. It is noteworthy that in the mass spectrum of all compounds, two peaks with higher intensity were observed: one representing an acylium ion formed during fragmentation, and the other corresponding to the molecular ion. In contrast to substrates 3a-3f, where the molecular ion peak predominated, the acylium ion peak was notably higher in intensity for compounds 4a-4d and 4f. This heightened intensity of the acylium ion serves as another indication of α,β-bond reduction, which favors easy fragmentation during the process. Furthermore, FTIR spectra of the reduced products 4a-4d and 4f exhibited a distinct band at 2924 cm-1, absent in the spectra of compounds 3a-3f. These distinctive bands correspond to the symmetrical and asymmetrical stretching of the bond between the two sp3 carbons, originating from the biocatalytic reduction catalyzed by P. citrinum CBMAI 1186. The precise position of the biocatalytic reduction in compounds 4a-4d and 4f was verified through the analysis of 1H NMR spectrum. This spectrum revealed two distinct signals: a triplet and a quartet, corresponding to the methylene hydrogens bonded to Cα and Cβ (Figure 3). These signals indicate sp3 hybridization due to biocatalytic reduction. Additionally, two methine hydrogen signals were detected, specifically those associated with Cγ and Cδ, with observed J values ranging from 12 to 16 Hz. This range suggested the E-configuration for the remaining double bond in the structure. The regioselectivity of the enzyme was evident, demonstrating the selective reduction of the α,β-bond.
In the 13C NMR spectrum (100 MHz, CDCl3), the most unshielded signals were assigned to the carbon atoms adjacent to the carbonyl group. The 13C NMR spectra for all compounds exhibited a total of 13 peaks, indicating 15 carbon atoms with sp2-type hybridization and 2 carbon atoms with sp3-type hybridization. The selective reduction of the α,β-bond from α,β,γ,δ-unsaturated ketones using chemical catalysts has been previously reported in the literature. Ranu and Samanta14 demonstrated this reduction using a borohydride and indium(III) chloride (InCl3) solution in anhydrous acetonitrile. Zhan et al.12 reported the reduction of the α,β,γ,δ-unsaturated ketones employing tris(pentafluorophenyl)borane (B(C6F5)3) as a catalyst and triethylsilane (Et3SiH) as a reductant with hexafluoro-2-propanol (HFIP). Both studies achieved yields exceeding 90%. In our study, the highest yield attained was 74% for compound 4c using friendly reaction conditions. Although the chemical reduction process can present high selectivity and yield, the use of P. citrinum CBMAI 1186 has shown to be an attractive alternative. This approach involves a simple procedure where reactions with several compounds can be prepared under mild conditions using a stock of cells, without requiring the addition of catalysts, reducing agents or toxic and expensive solvents. Whole cells perform the reactions efficiently, offering numerous possibilities.
CONCLUSIONS This study demonstrated an efficient biocatalytic approach using whole cells of P. citrinum CBMAI 1186 for the regioselective reduction of α,β,γ,δ-unsaturated synthetic compounds (3a-3f). After six days, conversion rates for all compounds were close to or exceeded 90%. Notably, substrates containing fluorine and methyl groups exhibited high conversions (> 80%) within just 1-3 days. The carbonyl group remained intact in all reactions. The products were successfully purified, obtained in satisfactory yields, and characterized by NMR, FTIR, and X-ray analyses. The results suggest that substrate structure plays a more significant role than electronic effects in enzyme-substrate interactions. Ene-reductases present in P. citrinum CBMAI 1186 were likely responsible for the reductions observed. This work confirms the viability of using a simple, low-cost fungal-based system to promote regioselective reduction under mild conditions. Even when compared to isolated enzymes, whole-cell catalysis yielded promising results, highlighting its potential for α,β,γ,δ-unsaturated and α,β-unsaturated compounds. Further studies exploring additional substrates could provide deeper insights into the catalytic properties of the enzyme, reinforcing its applicability in biocatalysis.
SUPPLEMENTARY MATERIAL Supplementary material, chromatograms (GC-MS) for the reactions performed, as well as the 1H and 13C NMR spectra and infrared spectra for the characterization of the starting material and the obtained products, are available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
DATA AVAILABILITY STATEMENT All data are available in the text and supplementary material.
ACKNOWLEDGMENTS P. H. Damada thanks to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES, 88887.372100/2019-00) - finance code 001. This study was also financed in part by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) projects 2019/07654-2, 2016/20155-7, 2021/10066-5, 2018/15904-6 (A. L. M. Porto), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) project 302528/2017-2 (A. L. M. Porto) and 312505/2021-3 (J. Ellena). This research used facilities of the Brazilian Synchrotron Light Laboratory (LNLS), part of the Brazilian Center for Research in Energy and Materials (CNPEM), a private non-profit organization under the supervision of the Brazilian Ministry for Science, Technology, and Innovations (MCTI). The MANACÁ beamline staff, especially Dr. A. F. Z. Nascimento is acknowledged for the assistance during the experiments (proposal number 20231693).
REFERENCES 1. Hughes, G.; Lewis, J. C.; Chem. Rev. 2018, 118, 1. [Crossref] 2. Sheldon, R. A.; Woodley, J. M.; Chem. Rev. 2018, 118, 801. [Crossref] 3. Atalah, J.; Cáceres-Moreno, P.; Espina, G.; Blamey, J. M.; Bioresour. Technol. 2019, 280, 478. [Crossref] 4. de Carvalho, C. C. C. R.; Microb. Biotechnol. 2017, 10, 250. [Crossref] 5. Lin, B.; Tao, Y.; Microb. Cell Fact. 2017, 16, 106. [Crossref] 6. Wu, S.; Snajdrova, R.; Moore, J. C.; Baldenius, K.; Bornscheuer, U. T.; Angew. Chem., Int. Ed. 2021, 60, 88. [Crossref] 7. Birolli, W. G.; Lima, R. N.; Porto, A. L. M.; Frontiers in Microbiology 2019, 10, 1453. [Crossref] 8. Hyde, K. D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A. G. T.; Abeywickrama, P. D.; de Silva, N. I.; Doilom, M.; Karunarathna, S. C.; Luangharn, T.; Mortimer, P. E.; Wei, D.; Fungal Diversity 2019, 97, 1. [Crossref] 9. Borges, K. B.; Borges, W. D. S.; Durán-Patrón, R.; Pupo, M. T.; Bonato, P. S.; Collado, I. G.; Tetrahedron:Asymmetry 2009, 20, 385. [Crossref] 10. Ye, M.; Ye, Y.; Du, Z.; Chen, G.; Bioprocess Biosyst. Eng. 2021, 44, 1003. [Crossref] 11. Toogood, H. S.; Scrutton, N. S.; ACS Catal. 2018, 8, 3532. [Crossref] 12. Zhan, X. Y.; Zhang, H.; Dong, Y.; Yang, J.; He, S.; Shi, Z. C.; Tang, L.; Wang, J. Y.; J. Org. Chem. 2020, 85, 6578. [Crossref] 13. Cong, Y.; Zeng, X.; Chin. J. Org. Chem. 2020, 40, 2411. [Crossref] 14. Ranu, B. C.; Samanta, S.; J. Org. Chem. 2003, 68, 7130. [Crossref] 15. Kabsch, W.; Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125. [Crossref] 16. Sheldrick, G. M.; Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3. [Crossref] 17. Sheldrick, G. M.; Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3. [Crossref] 18. Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H.; J. Appl. Crystallogr. 2009, 42, 339. [Crossref] 19. Macrae, C. F.; Sovago, I.; Cottrell, S. J.; Galek, P. T. A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G. P.; Stevens, J. S.; Towler, M.; Wood, P. A.; J. Appl. Crystallogr. 2020, 53, 226. [Crossref] 20. Corrêa, R.; Fenner, B. P.; Buzzi, F. C.; Cechinel Filho, V.; Nunes, R. J.; Z. Naturforsch., C: J. Biosci. 2008, 63, 830. [Crossref] 21. Jin, H.; Xiang, L.; Wen, F.; Tao, K.; Liu, Q.; Hou, T.; Ultrason. Sonochem. 2008, 15, 681. [Crossref] 22. Polaquini, C.; Torrezan, G.; Santos, V.; Nazaré, A.; Campos, D.; Almeida, L.; Silva, I.; Ferreira, H.; Pavan, F.; Duque, C.; Regasini, L.; Molecules 2017, 22, 1685. [Crossref] 23. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Vyvyan, J. R.; Introduction to Spectroscopy, 5th ed.; Cengage Learning: Stamford, 2015. 24. Corrêa, M. J. C.; Nunes, F. M.; Bitencourt, H. R.; Borges, F. C.; Guilhon, G. M. S. P.; Arruda, M. S. P.; Marinho, A. M. R.; Santos, A. S.; Alves, C. N.; Brasil, D. S. B.; Santos, L. S.; J. Braz. Chem. Soc. 2011, 22, 1333. [Crossref] 25. Filippucci, S.; Tasselli, G.; Labbani, F. Z. K.; Turchetti, B.; Cramarossa, M. R.; Buzzini, P.; Forti, L.; Fermentation 2020, 6, 29. [Crossref] 26. Tseng, N.; Wang, N.; Szostek, B.; Mahendra, S.; Environ. Sci. Technol. 2014, 48, 4012. [Crossref] 27. Chen, Q.; Liu, J.; Zhang, H.; He, G.; Fu, M.; Enzyme Microb. Technol. 2009, 45, 175. [Crossref] 28. Damada, P. H.: Descoberta e Exploração Biocatalítica de Ene-Redutases Fúngicas; Tese de Doutorado, Universidade de São Paulo, São Carlos, Brasil, 2024. [Link] accessed in May 2025 29. Parmeggiani, F.; Brenna, E.; Colombo, D.; Gatti, F. G.; Tentori, F.; Tessaro, D.; ChemBioChem 2021, 23, e202100445. [Crossref] 30. Roy, T. K.; Sreedharan, R.; Ghosh, P.; Gandhi, T.; Maiti, D.; Chem. - Eur. J. 2022, 28, e202103949. [Crossref] 31. Fan, X. Y.; Yu, Y.; Yao, Y.; Li, W. D.; Tao, F. Y.; Wang, N.; J. Agric. Food Chem. 2024, 72, 18305. [Crossref] 32. Ferreira, I. M.; Meira, E. B.; Rosset, I. G.; Porto, A. L. M.; J. Mol. Catal. B: Enzym. 2015, 115, 59. [Crossref] 33. Aalbers, F. S.; Fraaije, M. W.; ChemBioChem 2019, 20, 51. [Crossref] 34. Ferreira, I. M.; Fiamingo, A.; Campana-Filho, S. P.; Porto, A. L. M.; Mar. Biotechnol. 2020, 22, 348. [Crossref]
Guest Editor handled this article: Ivo José C. Vieira "A ciência avança devido à dedicação daqueles que estudam a natureza". |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access